Notes
Genbank
Many entries will not have all the data listed above - for example the gene ID may be missing (e.g. entry AB000381).
You should only consider entries that include CDS records - i.e. contain a coding sequence.
GenBank will also contain other oddities: several CDSs per Genbank locus (just use the first one); alternative splicing (just use the first one); reverse complement sequences (you need to reverse complement it yourself...); gene names not unique (just use the first one).
The GenBank format is described at http://www.ncbi.nlm.nih.gov/Sitemap/samplerecord.html and at http://www.insdc.org/files/feature_table.html. Pay particular attention to the 'FEATURES' section and, within that, the CDS (coding sequence) records. Note that a particular record may contain more than one coding sequence.
A small percentage of coding sequences require you to join stretches of DNA from another entry to form the complete coding sequence. (e.g. the coding sequence contains bases 28199-28271 and 28881-28988 from one entry and bases 1003-1983 from another entry). For the purpose of this exercise, these should simply be omitted from the database.
Mutations, variations, and alternative splicing should all be ignored. (In the case of alternative splicing, just take the first splice variant.)
Codon Usage
A typical codon usage table is shown here (reproduced from http://www.sci.sdsu.edu/~smaloy/MicrobialGenetics/topics/in-vitro-genetics/codon-usage.gif).
For the non-Biologists, the genetic code is redundant. For most amino acids, there are 4 codons that will all code the same amino acid. In the example table, you will see, in the bottom right, the 4 codons for Glycine. For each codon, the table shows the percentage of all codons that this codon represents (for example, GGU is 2.8% of all codons - often the fraction of 1000 codons is given instead, so this would be 28). In addition the ratio is shown - i.e. for the 4 codons that encode Glycine, GGU is used 38% of the time, GGC 40%, GGA 9% and GGG 13%. This relative usage of different codons is species-specfic.
In general, any given gene will approximately reflect this ratio of codon usage. Expressing a human gene in E. coli may lead to poor expression because the codon usage in E. coli is different from that in humans and the E. coli translation apparatus cannot translate its rare codons as efficiently.
Sometimes there is selective pressure for a protein to use rare codons. Because these are translated more slowly, it causes the translation apparatus to stall or slow down allowing the part of the protein that has already been synthesized to fold.
What we are looking for in the web interface is the ability to display the codon usage frequencies for the coding regions of a particular entry. In addition we want to be able to compare this with the overall codon usage frequencies in the whole chromosome. (You would clearly need to pre-calculate this from the data in your database.)
How you decide to display this information is up to you, but a simple thing would be to provide a table of the form shown here for a particular entry together with the table for the whole chromosome. Alternatively, you might combine the data into one table. You might use colour to indicate codons that are over- or under-used (perhaps you could work out some statistics to see if they are significantly over- or under-used). You might also want to use colour to annotate your sequence by highlighting uncommon codons. Think about what might be useful to your end-user.
Restriction Enzymes
Restriction Enzymes are enzymes that can cut DNA at specific sequence locations - generally these are palindromic. Because they leave 'sticky' ends after they cut, they are the fundamental tools of molecular biology - if you cut a piece of DNA out from one place, you can splice it into another place (for example a vector - a virus that infects bacteria - that allows the DNA to be expressed as protein in a bacterium). This is the idea behind 'cloning' a gene.
So the biological desire here is to find one or more restriction enzymes that would allow us to cut the gene out of the human DNA and splice it into some other piece of DNA that allows it to be handled in the lab. We want to find restriction enzymes that cut before and/or after the coding regions of the gene itself, but not WITHIN the coding regions - if we cut within the coding regions we would mess up the important part of the DNA. The 5' region is the part before the coding sequence and the 3' region is the part after the coding sequence.
For the biologists... In reality we would probably want to do this with a cDNA library not with genomic DNA, so that introns were not included. Remember this is a computing exercise!
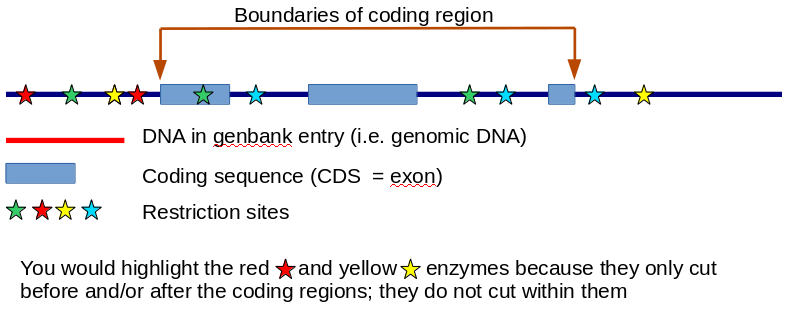
So the idea is simply to identify all the sites at which each restriction enzyme cuts. If a particular enzyme cuts before and/or after the coding regions, but not within the coding regions, then we highlight it in colour or whatever (maybe just with some text to say 'this is a good enzyme!') to say that this would be a useful enzyme to use for the genetic manipulation of this gene.
You should download some general guidance on what is expected. See the Good Code section for further help.