Part 1: Protein Structure - Backbone torsion angles
AIM: To ensure you understand what torsion angles are and how they are measured.
A torsion angle (or 'dihedral' angle) is the 'twist' angle along a bond:
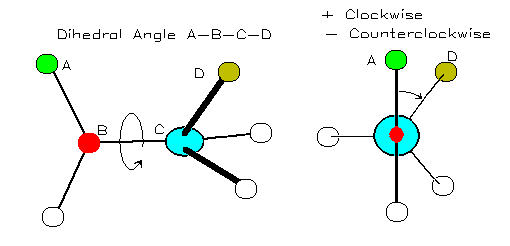
Image from http://bmbiris.bmb.uga.edu/wampler/tutorial/prot2.html
The torsion angle is positive when the atom farther away (atom D in the diagram) moves clockwise with respect to the closer atom (atom A in the diagram).
The protein backbone can be described in terms of the phi, psi and omega torsion angles of the bonds:
- The phi angle is the angle around the -N-CA- bond (where 'CA' is the alpha-carbon)
- The psi angle is the angle around the -CA-C- bond
- The omega angle is the angle around the -C-N- bond (i.e. the peptide bond)

Image from http://bmbiris.bmb.uga.edu/wampler/tutorial/prot2.html
First, download this peptide. To download it, you must right click the link and choose 'Save Target As...' from the menu.
Either close and restart PyMol, or select 'Reinitialize' (and 'Everything' if present) from the 'File' menu.
Now view the peptide using PyMol. You will see a fragment from the protein crambin with the sequence Thr-Gly-Cys-Ile-Ile.
- Load the pept.pdb PDB file of the peptide into PyMol
- Set the view to a wireframe rendering, by clicking 'S' next to 'all' in the display panel, hover over 'as', move to the other menu and click 'wire'
- Look at the peptide. Identify the backbone nitrogens (coloured in blue) and the backbone carbonyl groups (red sticks on the side of the green backbone)
- Work your way along the backbone and identify each C-alpha atom and its sidechain
- Now in the Display panel, next to 'all', click 'L', then click 'atom name'
- Click the black background to remove the pink squares
- You will now see each atom has a label next to it
Having identified where the peptide bonds are, we will have a look at their torsion angles - since there are no prolines, you would expect these to be trans. Let's see if they are:
- At the bottom right of the main window, you should see 'Selecting Residues'. Click this multiple times until it says 'Selecting Atoms'
- Click on the black background to ensure there are no pink squares
- Pressing Control and using the middle mouse button, click in sequence a 'CA', 'C', 'N', 'CA' (of the next amino acid). If you don't have a 3-button mouse, you can also do a double right-button click on each atom in turn. More help
- In the command window, type 'dihedral'
- A yellow or grey arc will appear on the screen with a dihedral angle displayed next to it. This should be close to 180 or -180 (within 10 degrees).
- Remove the highlighting of the CA atoms, by Ctrl-middle-clicking on the black background (or double-click the right mouse button). More help
Rather than doing this for every dihedral, we will simply look at the bonds visually to see if they are close to 180 degrees.
First we will highlight the peptide bonds:
- In the command window, type: select name N+C
- In the command window, type: show sticks, sele
Now we will show the C-alpha atoms as spheres
- In the command window, type: sele name ca
- In the command window, type: show spheres, sele
You will see the 'CA' atoms highlighted with huge spheres! - In the command window, type: alter sele, vdw=0.5
- In the command window, type: rebuild
Finally we will look at each peptide bond in turn:
- Now rotate the view so that each peptide bond in turn is orientated such that it is coming out of the screen. (i.e. the line of the N-C bond is viewed end-on and looks like a small circle).
- You should see that the bonds to the adjacent C-alpha atoms (which are now shown as spheres) are at approximately 180 degrees to one another.
Repeat what you did above, to calculate the omega (peptide bond) torsion angle between Thr30 and Gly31. Record this value.
Record the omega (peptide bond) torsion angle between Gly31 and Cys32.